In a new paper, published in the Proceedings of the National Academy of Sciences (PNAS), we examine the gap between the global emissions of HFCs that we infer from atmospheric observations and those reported by developed countries. The findings of the paper are summarised in a University of Bristol press release:
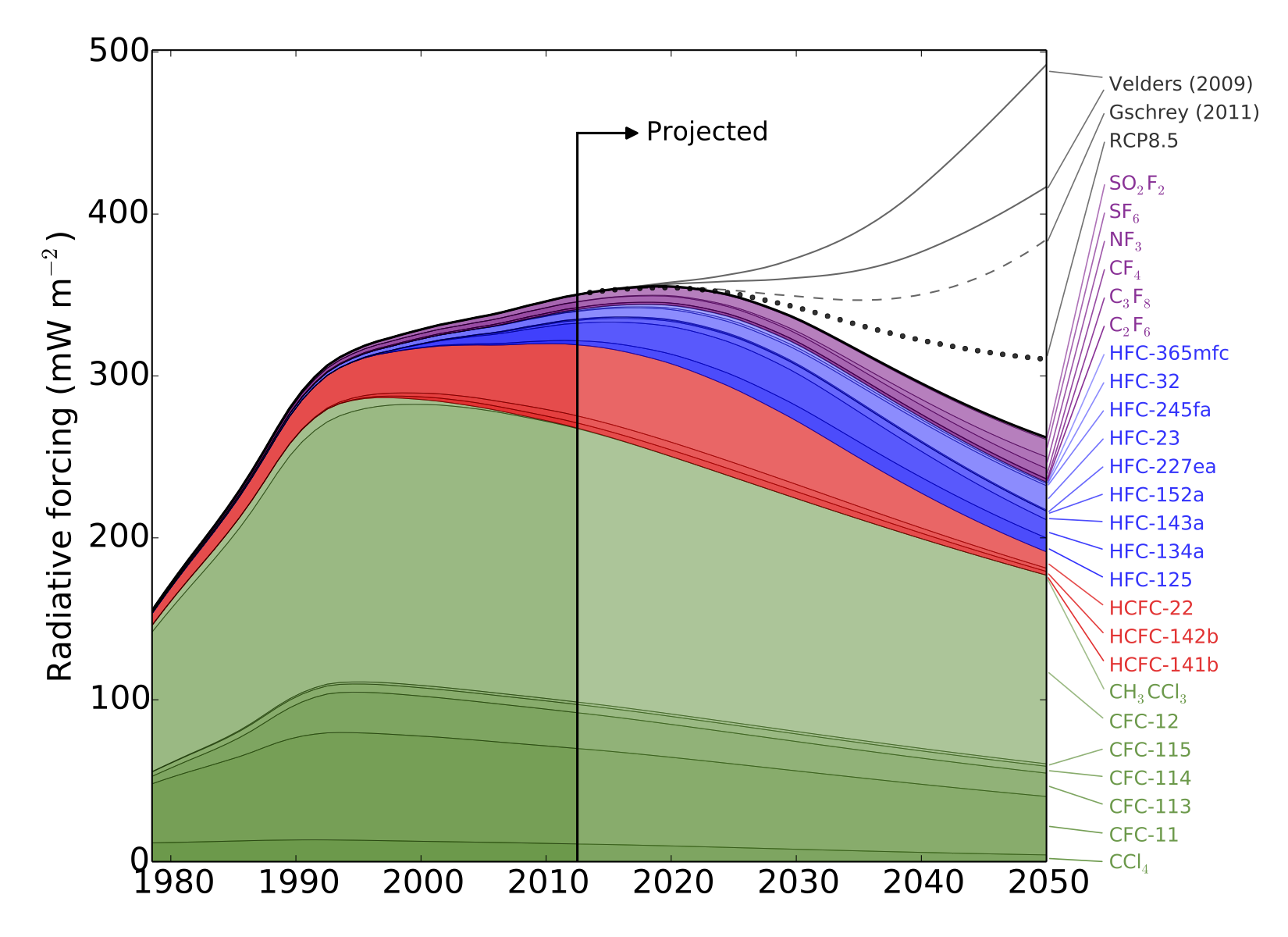
Until now, there has been little verification of the reported emissions of hydrofluorocarbons (HFCs), gases that are used in refrigerators and air conditioners, resulting in an unexplained gap between the amount reported, and the rise in concentrations seen in the atmosphere. This new study shows that this gap can be almost entirely explained by emissions from developing countries.
Currently only 42 countries are required to provide detailed annual reports of their emissions to the United Nations Framework Convention on Climate Change (UNFCCC).
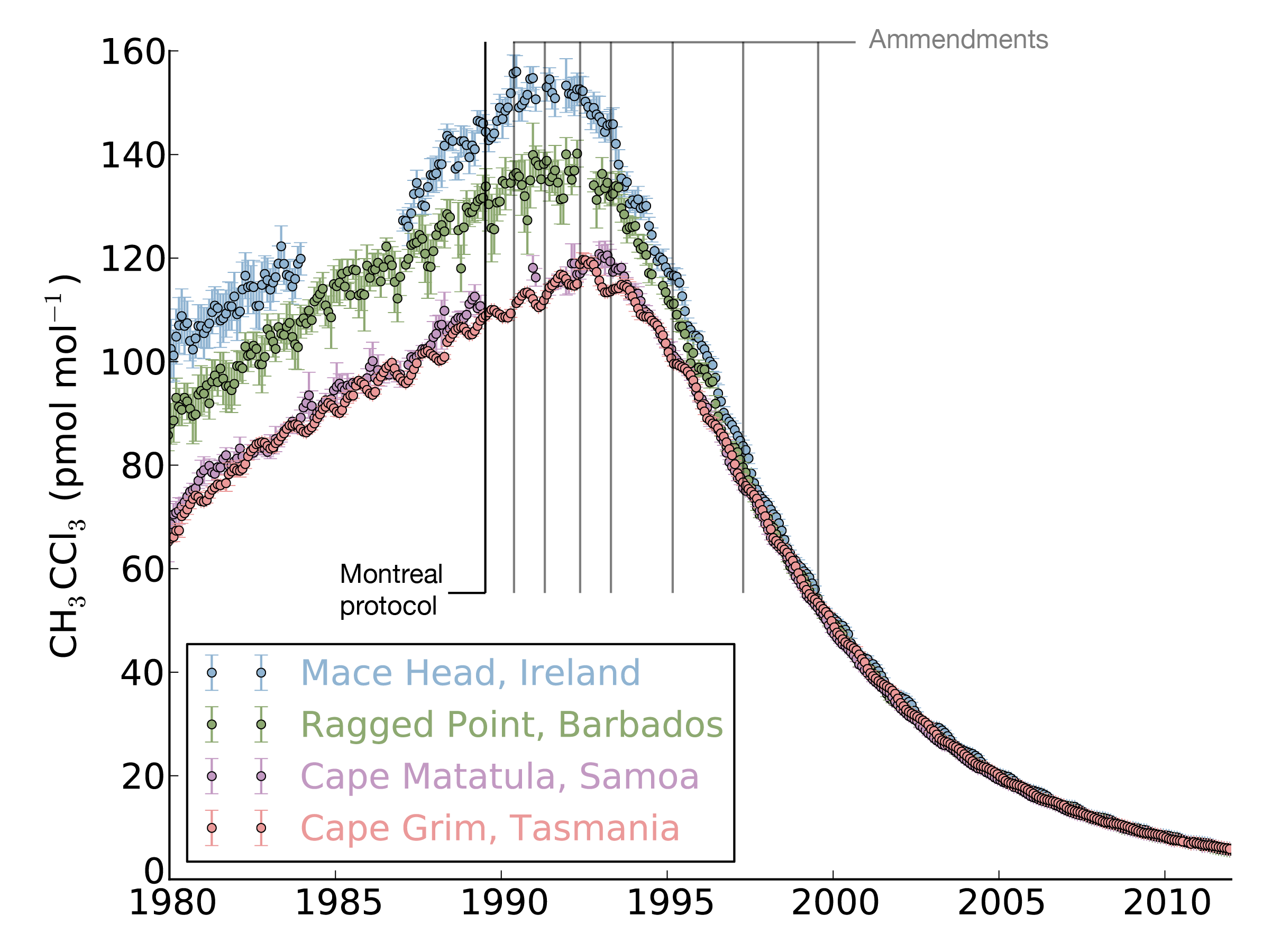
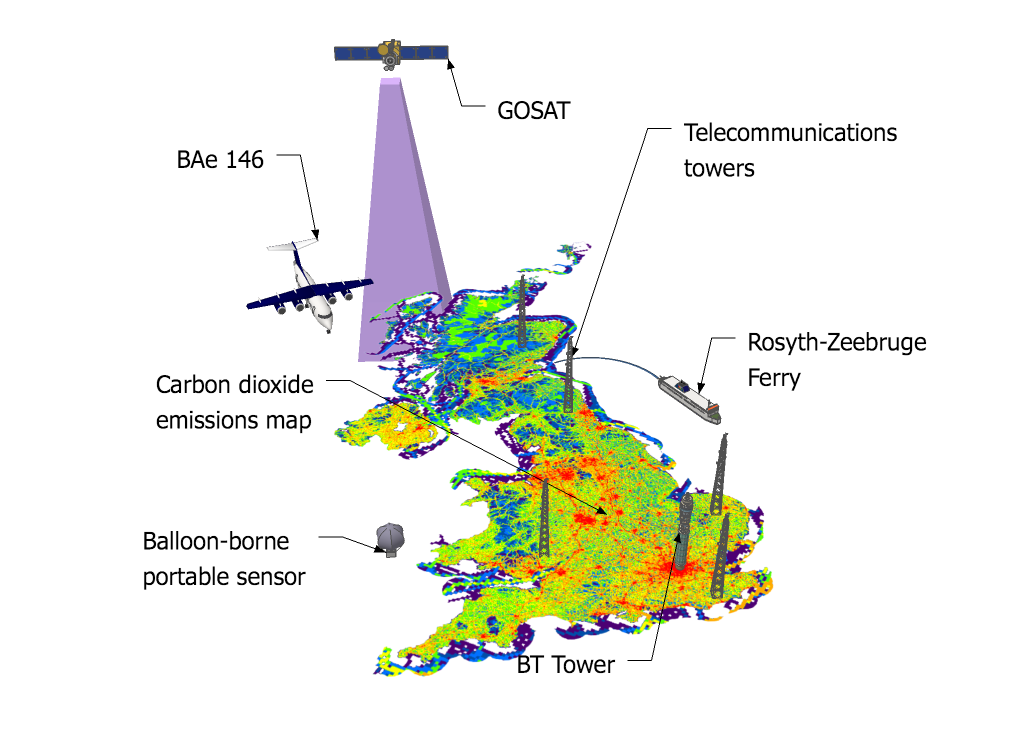
The study, led by Mark Lunt from Bristol’s School of Chemistry used HFC measurements from the international Advanced Global Atmospheric Gases Experiment (AGAGE), in combination with models of gas transport in the atmosphere, to evaluate the total emissions that are reported to the UNFCCC each year.
HFCs are potent greenhouse gases; per tonne of emissions, each gas measured in this work is hundreds or even thousands of times more effective than carbon dioxide at trapping the radiation that warms the Earth.
There is currently no global agreement to regulate the emissions of these compounds, although proposals have been made to begin phasing out their use.
Mark Lunt said: “Any phase-out mechanism would likely be more stringent for the developed countries, but these results show that emissions from non-reporting countries are also highly significant.”
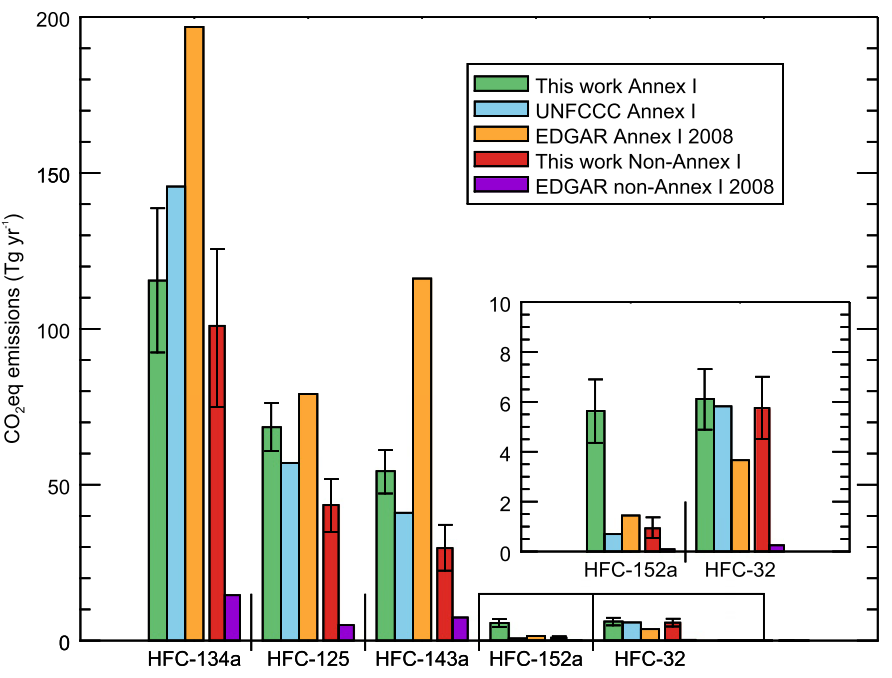
Meanwhile, the researchers note that although their estimates of total emissions from developed countries are broadly consistent with the reports that they compile, this does not necessarily mean that the emissions of each gas are being accurately reported.
In fact, the results suggest that the most commonly used HFC is significantly over-reported whilst some other HFCs are under-reported.
Dr Matt Rigby from the University of Bristol, who co-authored this work, said: “It appears as if the apparent accuracy of the aggregated HFC emissions from developed countries is largely due to a fortuitous cancellation of errors in the individual emissions reports.”
Professor Ron Prinn from the Massachusetts Institute of Technology (MIT), who leads the AGAGE network, added: “This study highlights the need to verify national reports of greenhouse gas emissions into the atmosphere. Given the level of scrutiny these reports are under at the moment, it is vitally important that we improve our ability to use air measurements to check that countries are actually emitting what they claim.”